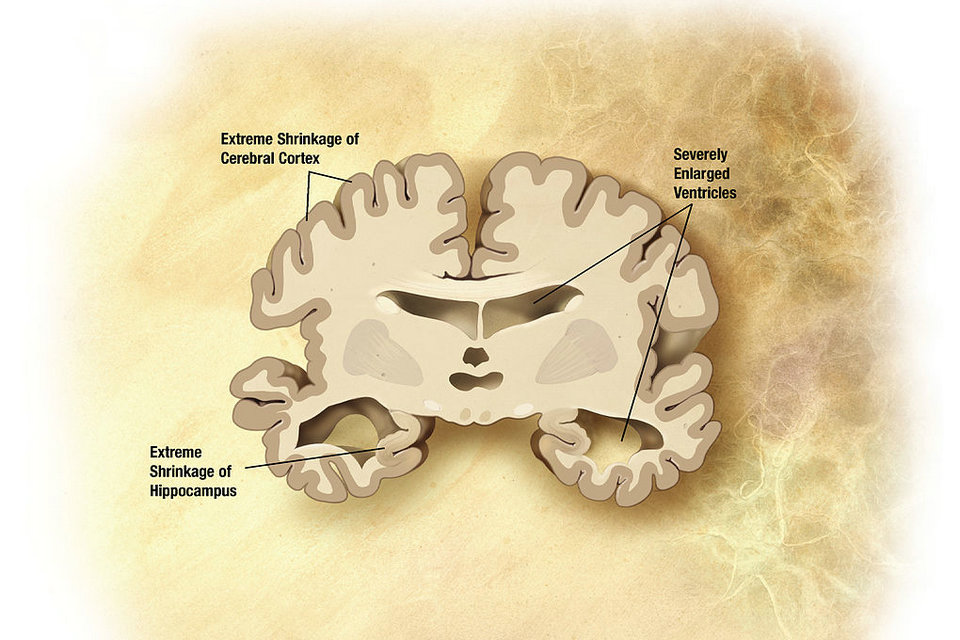
En avril 2014, 315 essais cliniques ouverts étaient en cours pour comprendre et traiter la maladie d’Alzheimer. 42 de ces études étaient des essais ouverts de phase trois chez l’homme, la dernière étape avant l’approbation et la commercialisation de la Food and Drug Administration (FDA) des États-Unis. Au cours de la décennie 2002-2012, 244 composés ont été évalués dans le cadre d’essais de phase I, II ou III, et un seul d’entre eux (la mémantine) a été approuvé par la FDA (bien que d’autres soient encore en attente).
Il y a différentes approches. Une approche consiste à réduire la bêta de l’amyloïde, par exemple avec le bapineuzumab, un anticorps dans les études de phase III pour les patients en stade léger à modéré; le semagacestat, un inhibiteur de la y-sécrétase, MPC-7869; et UB-311, ACC-001 ou CAD106, vaccins contre la bêta-amyloïde. D’autres approches sont les agents neuroprotecteurs, comme AL-108 (phase II terminée); ou atténuation de l’interaction métal-protéine, comme dans le cas de PBT2 (phase II terminée). Une autre approche consiste à utiliser des stimulants cognitifs généraux, comme cela peut être le cas pour la mémantine, un médicament approuvé aux États-Unis et dans l’Union européenne pour traiter les symptômes de la MA modérée à sévère. Chez les animaux, les ultrasons ont été utilisés pour pénétrer la barrière hémato-encéphalique et activer les cellules microgliales afin d’éliminer la bêta-amyloïde et de restaurer la fonction de mémoire. Enfin, il existe des études de base sur l’origine et les mécanismes de la maladie d’Alzheimer.
Traitements en développement clinique
Plusieurs traitements potentiels de la maladie d’Alzheimer sont à l’étude, y compris plusieurs composés à l’étude dans des essais cliniques de phase III. La recherche clinique la plus importante porte sur le traitement potentiel de la pathologie sous-jacente, pour laquelle la réduction de la bêta-amyloïde est une cible courante des composés à l’étude.
Immunothérapie à la bêta-amyloïde
L’immunothérapie ou la vaccination contre la maladie d’Alzheimer stimule le système immunitaire à attaquer la bêta-amyloïde. Une approche est l’immunisation active, ce qui stimulerait une réponse immunitaire permanente. Le vaccin AN-1792 s’est révélé prometteur dans les premiers essais sur la souris et chez l’homme, mais lors d’un essai de phase II en 2002, 6% des sujets (18 sur 300) ont développé une inflammation cérébrale grave ressemblant à une méningo-encéphalite. L’essai a été arrêté. Lors des suivis à long terme, 20% des sujets avaient développé des taux élevés d’anticorps anti-bêta-amyloïde. Alors que les patients sous placebo et ceux qui ne répondaient pas aux anticorps se détérioraient, ceux-ci présentaient un degré de stabilité des niveaux cognitifs évalué par la batterie de tests neuropsychologiques (bien que pas par d’autres mesures) et avaient des niveaux plus bas de protéine tau dans leur liquide céphalo-rachidien . Ces résultats peuvent suggérer une activité réduite de la maladie dans le groupe anticorps-répondeur. Les autopsies ont montré que la vaccination entraînait la disparition des plaques amyloïdes, mais n’empêchait pas la neurodégénérescence progressive.
Une étude de phase IIA de l’ACC-001, une version modifiée de l’AN-1792, recrute actuellement des sujets.
Un vaccin Aβ s’est avéré efficace contre la myosite à inclusions chez des modèles murins.
Immunothérapie passive
Également dérivé du programme d’immunothérapie AN-1792, il existe une approche par anticorps perfusée appelée vaccin passif en ce sens qu’elle n’invoque pas le système immunitaire et nécessiterait des perfusions régulières pour maintenir les niveaux d’anticorps artificiels. Les hémorragies micro-cérébrales peuvent constituer une menace pour ce processus.
Le bapineuzumab, un anticorps anti-amyloïde-β, était en cours de développement; Cependant, le médicament a échoué lors des essais cliniques de phase 3. L’anticorps a été conçu comme étant essentiellement identique à l’anticorps naturel déclenché par le vaccin AN-1792 antérieur.
Une étude récente a montré que des médicaments anticancéreux approuvés par la FDA, des inhibiteurs de la PD-1, pourraient être bénéfiques pour les patients atteints de la maladie d’Alzheimer. L’étude a utilisé un modèle murin de la maladie d’Alzheimer et un anticorps anti-PD-1 pour démontrer une réduction statistiquement significative des plaques de l’amyloïde-β et une performance cognitive améliorée.
Cibler la production de peptide bêta-amyloïde
Inhibition de la gamma sécrétase
La gamma sécrétase est un complexe protéique considéré comme un élément fondamental du développement du peptide bêta-amyloïde. Le semagacestat, un inhibiteur de la gamma sécrétase, n’a montré aucun bénéfice pour les patients atteints de la maladie d’Alzheimer au cours des essais cliniques.
Modulation de gamma sécrétase
Tarenflurbil (MPC-7869, anciennement R-flubiprofène) est un modulateur de la gamma sécrétase, parfois appelé agent réducteur sélectif de l’amyloïde bêta 42. On pense que la production de bêta-amyloïde toxique est réduite au profit de formes plus courtes du peptide. Des résultats négatifs ont été annoncés concernant le tarenflurbil en juillet 2008 et le développement ultérieur a été annulé.
Inhibition de la bêta sécrétase
En octobre 2015, Amgen et Novartis avaient conclu un accord de codéveloppement visant à faire progresser l’inhibiteur oral de la bêta-sécrétase CNP520.
Atténuation des interactions métal-protéine
PBT2 est une 8-hydroxy quinoléine qui élimine le cuivre et le zinc du liquide céphalorachidien, qui sont considérés comme des catalyseurs nécessaires à l’agrégation bêta de l’amyloïde. Ce médicament a fait l’objet d’un essai de phase II sur des patients atteints de la maladie d’Alzheimer au début de l’étude et a donné des résultats préliminaires prometteurs, mais non détaillés.
Statines
La simvastatine, une statine, stimule les cellules endothéliales vasculaires cérébrales afin de créer un éjecteur bêta-amyloïde. L’utilisation de cette statine peut avoir un lien de causalité avec une diminution du développement de la maladie.
Correction métabolique
Cette approche est basée sur l’aspect dominant de la maladie d’Alzheimer, commune à de nombreuses autres maladies neurodégénératives: le déficit énergétique. Il a d’abord été noté pour le cas d’insuffisance d’insuline dans le cerveau de patients atteints de la maladie d’Alzheimer. À cause de cela, la maladie d’Alzheimer a été appelée diabète de type 3 et les thérapies de modification de l’insuline sont en cours de traitement.
Autres produits pharmaceutiques
Plusieurs autres produits pharmaceutiques sont à l’étude pour traiter la maladie d’Alzheimer.
Allopregnanolone
L’allopregnanolone a été identifiée comme un agent médicamenteux potentiel. Les taux de neurostéroïdes, tels que l’alloprégnanolone, diminuent dans le cerveau chez les personnes âgées et atteintes de la maladie d’Alzheimer. Allopregnanolone a été montré pour aider la neurogenèse qui inverse les déficits cognitifs dans un modèle murin de la MA.
Bloqueurs des récepteurs de l’angiotensine
Une analyse rétrospective de cinq millions de dossiers de patients avec le système du département américain des anciens combattants a révélé que différents types de médicaments antihypertenseurs couramment utilisés avaient des résultats très différents pour la MA. Les patients prenant des antagonistes des récepteurs de l’angiotensine (ARA) étaient 35 à 40% moins susceptibles de développer une MA que ceux utilisant d’autres anti-hypertenseurs.
Antibiothérapie
Plusieurs études utilisant des antibiotiques dans des modèles animaux de la maladie d’Alzheimer ont indiqué que la minocycline et la doxycycline exerçaient un effet protecteur en empêchant la mort des neurones et en ralentissant l’apparition de la maladie.
Un essai préliminaire d’antibiothérapie à la doxycycline et à la rifampine menée à l’Université McMaster avait montré son efficacité pour retarder l’évolution de la maladie: « En conclusion, un traitement de 3 mois à base de doxycycline et de rifampine a réduit l’aggravation cognitive à 6 mois chez les patients atteints de MA légère à modérée. » Un réexamen des mêmes données en utilisant « l’analyse par la AUC de l’indice regroupé a montré un effet de traitement significatif sur la période de 12 mois ». Cependant, lorsque ces conclusions préliminaires ont été suivies d’un essai ultérieur plus important, ils ont constaté que « la doxycycline ou la rifampine, seules ou combinées, n’a aucun effet bénéfique sur la cognition ou la fonction de [maladie d’Alzheimer] ».
Thérapie antivirale
Une étude montrant la colocation du virus de l’herpès simplex avec des plaques amyloïdes suggère la possibilité que la MA puisse être traitée avec un médicament antiviral.
Cannabis
Peu de recherches ont été menées sur l’utilisation possible du cannabis pour lutter contre le déclin cognitif et la démence dans la maladie d’Alzheimer. Aucun résultat de grande qualité n’indique aucun bénéfice à partir de 2015.
Dimebon
En juillet 2008 également, les résultats d’une étude dans laquelle un antihistaminique, auparavant disponible en Russie, Dimebon, avait été administré à un groupe de patients atteints de la MA ont été annoncés. Le groupe recevant Dimebon s’est quelque peu amélioré au cours des six mois de l’étude (et cela s’est poursuivi pendant les six mois suivants), tandis que ceux du groupe placebo se sont détériorés. Malheureusement, les essais de phase III consécutifs n’ont pas montré d’effets positifs significatifs sur les critères d’évaluation principaux et secondaires. En mars 2010, les promoteurs ont reconnu que les premiers résultats de l’essai de phase III montraient que, si le médicament avait été bien toléré, ses résultats ne différaient pas significativement de ceux du groupe placebo.
Étanercept
L’étanercept est à l’étude dans la maladie d’Alzheimer. Son utilisation est controversée.
Sensibilisants à l’insuline et insuline intranasale
Des études récentes suggèrent un lien entre la résistance à l’insuline et la MA (la sensibilité des cellules adipeuses à l’insuline peut diminuer avec le vieillissement): lors des essais cliniques, un certain sensibilisateur à l’insuline appelé « rosiglitazone » a amélioré la cognition chez un sous-groupe de patients atteints de MA; in vitro, les effets bénéfiques de la rosiglitazone sur les neurones corticaux primaires du rat ont été démontrés. Les recherches initiales suggèrent que l’insuline intranasale, augmentant les niveaux d’insuline dans le cerveau avec une augmentation minimale de l’insuline dans le reste du corps, pourrait également être utilisée. Des études précliniques montrent que l’insuline élimine du cerveau la bêta-amyloïde soluble quelques minutes après une injection systémique chez des souris transgéniques diabétiques modélisant la maladie d’Alzheimer.
Chlorure de méthylthioninium
En juillet 2008, des chercheurs ont annoncé des résultats positifs du chlorure de méthylthioninium (MTC), (nom commercial: Rember), un médicament qui a dissous les polymères de Tau. Les résultats de la phase II indiquent que c’est le premier traitement qui réussit à modifier l’évolution de la maladie dans la MA légère à modérée.
Récepteurs Sigma
Considéré à l’origine comme une protéine énigmatique, le récepteur sigma-1 a été identifié comme un chaperon moléculaire unique régulé par un ligand dans le réticulum endoplasmique des cellules.Cette découverte a conduit à l’examen de nombreux rôles proposés de ce récepteur dans de nombreuses maladies neurologiques, y compris la maladie d’Alzheimer.
Protéine de translocateur
Une étude réalisée en 2013 a montré que la protéine du translocateur peut prévenir et traiter partiellement la maladie d’Alzheimer chez la souris.
Agonistes TrkB
R7 est un précurseur médicamenteux de la 7,8-dihydroxyflavone, un agoniste de TrkB, le principal récepteur du facteur neurotrophique dérivé du cerveau (BDNF). R7 est actuellement en développement préclinique pour le traitement de la maladie d’Alzheimer.
Médicaments candidats modificateurs de la maladie
Candidats modificateurs de la maladie dans les essais cliniques de dernière phase sur la maladie d’Alzheimer
| Cible / approche | Notes (théoriques) | nom du candidat | Phase d’essai | Date de début de l’essai | Date de fin prévue | Inscription prévue | Population AD ciblée (gravité) | Population AD ciblée (génétique) | commentaires |
|---|---|---|---|---|---|---|---|---|---|
| Gamma Secretase Modulator / NSAID | Déplace la production bêta-amyloïde en espèces plus courtes et moins toxiques.Cible la γ-sécrétase. | R-flurbiprofène (Flurizan, MPC-7869) | Phase III (terminée) | Février 2005 | Mai 2008 | 1600 | Doux | n / a | Myriad Genetics a conclu que le médicament n’améliorait pas la capacité de réflexion ni la capacité des patients de mener à bien leurs activités quotidiennes de manière significative par rapport aux patients du groupe placebo.Ils ont annoncé le 30 juin 2008 qu’ils ne développeraient plus le médicament. |
| Inhibiteur de gamma sécrétase | Inhibe la gamma sécrétase, qui réduit les niveaux de bêta-amyloïde | Semagacestat (LY450139) | Phase III (terminée) | Septembre 2008 | Avril 2011 | 1 100 | Légère à modérée | n / a | Le 17 août 2010, Eli Lilly a annoncé son intention « d’arrêter le développement du semagacestat » car « il ne ralentissait pas la progression de la maladie et était associé à une détérioration … de la cognition et de la capacité à effectuer des activités de la vie quotidienne ».En outre, il est « associé à un risque accru de cancer de la peau ». |
| Anticorps à la bêta-amyloïde | Mimics anticorps naturel déclenché par AN-1792 | Bapineuzumab (aab-001) | Phase III (terminée) | Décembre 2007 | Avril 2012 | 1 121 | Légère à modérée | Apolipoprotéine E4 Porteurs uniquement | Le 6 août 2012, Pfizer et Johnson & Johnson ont annoncé qu’ils mettaient fin au développement d’une formulation intraveineuse de bapineuzumab.Les essais de phase III « n’ont montré aucun effet thérapeutique sur les résultats cognitifs ou fonctionnels. Les analyses de biomarqueurs ont montré que le bapineuzumab avait atteint sa cible, mais n’avait aucun bénéfice ». |
| Anticorps à la bêta-amyloïde | Mimics anticorps naturel déclenché par AN-1792 | Bapineuzumab (aab-001) | Phase III (terminée) | Décembre 2007 | Juin 2012 | 1 331 | Légère à modérée | Apolipoprotéine E4 non porteuses uniquement | Le 6 août 2012, Pfizer et Johnson & Johnson ont annoncé qu’ils mettaient fin au développement d’une formulation intraveineuse de bapineuzumab.Les essais de phase III « n’ont montré aucun effet thérapeutique sur les résultats cognitifs ou fonctionnels. Les analyses de biomarqueurs ont montré que le bapineuzumab avait atteint sa cible, mais n’avait aucun bénéfice ». |
| Atténuation de l’interaction métal-protéine | Les cibles primaires sont le cuivre et le zinc. Élimine le cuivre et le zinc du liquide céphalo-rachidien. | PBT2 (8-hydroxy quinoléine) | Phase II (terminée) | Décembre 2006 | Décembre 2007 | 80 | Maladie d’Alzheimer précoce | n / a | « N’a pas atteint son objectif principal d’une réduction statistiquement significative des niveaux de plaques de bêta-amyloïde chez les patients atteints de maladie d’Alzheimer au cerveau prodromique / légère. » »Aucune amélioration n’a été observée sur les critères secondaires d’activité métabolique, cognitive et fonctionnelle du cerveau; toutefois, la tendance était à la préservation du volume cérébral de l’hippocampe ». »Plus précisément, il y avait moins d’atrophie par rapport au groupe placebo. » |
| Fibrilisation de la bêta-amyloïde | Décompose les fibrilles neurotoxiques, permettant ainsi aux peptides amyloïdes de se nettoyer au lieu de former des plaques amyloïdes. | ELND005 (AZD-103, scyllo-inositol) | Phase II (terminée) | Décembre 2007 | Mai 2010 | 353 | Légère à modérée | n / a | Les résultats de la phase I étaient encourageants d’ici à août 2007. En décembre 2009, Elan et Transition ont annoncé conjointement que l’étude de phase II avait été modifiée de sorte que seule la dose de 250 mg deux fois par jour soit maintenue en raison du « taux plus élevé d’effets indésirables graves, y compris neuf « décès », dans les groupes recevant la dose la plus élevée (1 000 mg et 2 000 mg administrés deux fois par jour).Il a reçu la désignation fast track de la FDA américaine. |
| Neuroprotection | Peptide neuroprotecteur intra-nasal | AL-108 | Phase II (terminée) | Janvier 2007 | Jan 2008 | 120 | Déficience cognitive légère | n / a | Considéré comme un succès;Phase III pour commencer[douteux] |
| Inhibiteur de l’apoptose des cellules du cerveau | Fonctionne à travers de multiples mécanismes: bloque l’action des protéines bêta-amyloïdes neurotoxiques et inhibe les canaux calciques de type L, module l’action des récepteurs du glutamate AMPA et NMDA, peut exercer un effet neuroprotecteur en bloquant une nouvelle cible impliquant des pores mitochondriaux et en bloquant une Nombre d’autres récepteurs, y compris α-adrénergiques, 5-HT 2C , 5-HT 5A et 5-HT 6 | latrepirdine (nom commercial: Dimebon) | Phase II (terminée) | Sep 2006 | Nov 2007 (actuel) | 183 | Légère à modérée | n / a | En mars 2010, Pfizer a annoncé que l’essai de phase III CONNECTION n’avait pas permis d’atteindre ses objectifs principaux et secondaires.En janvier 2012, il a été annoncé que l’étude de phase III CONCERT ne respectait pas ses critères d’évaluation principaux.Les essais CONTACT et CONSTELLATION ont été terminés.Medivation et Pfizer ont cessé le développement de dimebon et ont donc décidé de mettre fin à leur collaboration en matière de co-développement et de marketing. |
| Anticorps naturels contre l’amyloïde bêta | source de plasma humain limite l’offre | IgIV | Phase II (terminée) | Février 2006 | Juin 2007 | 24 | Légère à modérée | n / a | Considéré comme un succès;Phase III pour commencer |
| Vaccin contre la bêta-amyloïde | Injecte la bêta-amyloïde modifiée (vaccin actif) | ACC-001 | Phase II | Nov 2007 | Mars 2012 | 228 | Légère à modérée | n / a | Suite du célèbre essai de vaccin AN-1792 |
Biomarqueurs non imageurs
Le biomarqueur le plus largement utilisé est le peptide bêta-amyloïde 1-42 (Aβ42), qui est mesuré dans le liquide céphalorachidien. Les niveaux de Aβ42 sont en accord avec la tomographie par émission de positrons (PET) bêta-amyloïde et les deux méthodes permettent de détecter la DA prodromique avec une précision tout aussi élevée.
Des études ont également montré que les personnes atteintes de MA présentaient une diminution du glutamate (Glu) ainsi que des ratios Glu / créatine (Cr), Glu / myo-inositol (mI), Glu / N-acétylaspartate (NAA) et NAA / Cr diminués par rapport à personnes normales. Les diminutions de NAA / Cr et de glutamate de l’hippocampe peuvent être un indicateur précoce de la MA.
Les premières recherches sur une petite cohorte de patients atteints de la maladie d’Alzheimer ont peut-être permis d’identifier des marqueurs d’autoanticorps pour la MA. L’applicabilité de ces marqueurs est inconnue.
Une petite étude réalisée en 2011 sur l’homme a montré que le suivi des variations de la déshydroépiandrostérone (DHEA) dans le sang en réponse à un stress oxydatif pourrait être un test indirect utile: les sujets présentant une MCI ne présentaient pas de variation de la DHEA, contrairement aux contrôles sains.
Une étude réalisée en 2013 sur 202 personnes à l’Université de la Sarre en Allemagne a révélé que 12 microARN dans le sang étaient précis à 93% dans le diagnostic de la maladie d’Alzheimer.
Thérapie par ultrasons
Des résultats préliminaires positifs chez des rats dotés d’une technologie ultrasonore non invasive visant à nettoyer le cerveau des plaques amyloïdes ont été rapportés dans Science Translational Medicine. Une équipe australienne a décrit cette stratégie sous forme d’échographie par rayonnement dans le tissu cérébral. En oscillant à des fréquences élevées, les ondes sonores combinées à des microbulles dans le sang sont capables d’ouvrir la barrière hémato-encéphalique, diminuant ainsi les défenses cérébrales pendant quelques heures – un intervalle dans lequel elles stimulent l’activation des cellules microgliales du cerveau (et également, donner aux médicaments ou au système immunitaire l’accès au cerveau). L’équipe rapporte avoir observé une importante clairière dans les amas bêta-amyloïdes, modification attribuée aux cellules microgliales étant donné que leur fonction est fondamentalement liée à l’élimination des déchets; et restauration complète de la mémoire perdue et des fonctions cognitives chez 75% des souris sur lesquelles elles ont été testées, sans dommage concomitant du parenchyme cérébral (ni dans le tissu entourant les plaques bêta-amyloïdes, ni ailleurs). Les souris traitées auraient présenté des performances améliorées dans trois tâches de mémoire: un labyrinthe, un test permettant de reconnaître de nouveaux objets, et une de les rappeler aux endroits qu’elles devraient éviter. Sur la base de ces résultats, l’équipe envisage de commencer les essais avec des modèles animaux plus élevés, tels que les moutons et les singes, afin d’avoir des essais humains en cours en 2017.
Approche bioinformatique
Bien que les études in silico aient permis de mieux comprendre la maladie d’Alzheimer (MA) dans de nombreux domaines, ces méthodologies ont encore des limites, car les outils bioinformatiques sont faussés en faveur des données connues. Néanmoins, les conclusions tirées de l’utilisation d’outils et de bases de données bioinformatiques disponibles au public ont permis de découvrir de nouveaux traitements et de susciter de nouvelles questions pour faciliter le processus de recherche de remèdes contre la MA.
Pathogenèse et biomarqueurs
À partir des modèles d’expression génique obtenus dans des ensembles de données de microréseaux, il est possible de mettre en évidence une corrélation entre la physiologie cellulaire et les maladies. Les études de divergence (par exemple, les calculs de divergence de Jensen-Shannon qui interprètent la différence dans l’expression des gènes et la probabilité de profils de distribution) révèlent une différence dans la distribution de l’expression des gènes entre les cerveaux AD et les cerveaux de vieillissement normaux. C’est-à-dire que les gènes exprimés qui ont une corrélation négative avec le vieillissement normal du cerveau mais qui ont une corrélation positive avec le cerveau atteint de MA sont des biomarqueurs possibles pour le diagnostic et le traitement de la MA.La combinaison des gènes KEGG et PATHWAY studio, ATP5C1, COX6A1, NDUFV2, PLCB1 et PPP3CA est un métabolisme et des gènes liés aux mitochondries qui ont été démontrés dans des échantillons de AD. De plus, des dérégulations métaboliques telles que l’homéostasie du calcium et la signalisation de l’insuline ont également été identifiées comme contribuant à l’apparition de la MA.Les gènes associés à la signalisation du calcium et de l’insuline sont découverts à l’aide de GATHER (outil bioinformatique en ligne d’analyse des signatures génomiques). En fait, la dégradation de la signalisation à l’insuline et la MA ont été considérées comme liées à de nombreux niveaux. Les alignements fonctionnels des séquences protéiques (ClustalW, MUSCLE, par exemple) et l’analyse phylogénétique (Phylip, Mega, par exemple) montrent que l’acétylcholinestérase (AChE) et la butyrylcholinestérase (BChE) sont fortement liées dans ces deux maladies. Une augmentation de la BChE contribue à une altération du métabolisme des lipoprotéines et à une insensibilité à l’insuline, et est positivement corrélée à l’hypertension et au diabète dans les études de corrélation. L’AChE permet la stabilisation du neurotransmetteur, l’acétylcholine (ACh), l’une des cibles principales du traitement de la maladie d’Alzheimer. Cependant, une étude pharmacologique in silico récente a examiné l’interaction médicament-maladie a montré que les inhibiteurs de l’AChE n’étaient peut-être pas la réponse au traitement de la MA. Les inhibiteurs de la PKC, de l’ARG, de l’HDAC et de la GSK3 qui régulent l’homéostasie du calcium et la modification génétique du cycle cellulaire et de l’apoptose pourraient être les futures cibles du traitement de la MA.
La plasticité neuronale est un acteur clé de la fonction cognitive qui ne peut être ignorée lors de l’étude de la progression de la maladie. Des études de micropuces ont montré que NEFM, NEFL et SV2B sont fortement régulés négativement dans les échantillons prélevés chez des patients atteints de MA grave. NEFL est un gène de neurofilament dont il a été démontré qu’il était lié à l’hypotrophie des axones chez les motorneurones lors de la mutation. Cependant, il a été démontré que les deux neurofilaments (NEFL et NEFM) sont impliqués dans une maladie neurologique, Charcot-Marie-Tooth, au lieu de la MA, qui démontrent des liens inconnus possibles entre la MA et d’autres maladies neurologiques. Le SV2B est un autre gène régulé négativement dans la MA et il a été démontré qu’il est lié à la neurodégénérescence, en particulier l’exocytose synaptique régulée par le calcium. La régulation à la baisse des gènes responsables de la synapse neurale et de la neuroplasticité est liée à une autre famille de protéines qui a été associée à la pathogenèse de la MA, EGR (réponse de croissance précoce). Cet EGR est régulé par la voie FOXO1 (Forkhead Box O1) régulée à travers la voie PI3K / Akt, qui est répertoriée comme l’une des voies d’avenir pour les médicaments anti-AD. Ces découvertes utilisant des méthodes de calcul permettent la connexion de différentes études et facilitent la compréhension de la complexité de la maladie ainsi que l’orientation vers de nouveaux biomarqueurs possibles de la MA.
Pharmacologie
Les traitements actuels des symptômes de la MA sont les inhibiteurs de l’acétylcholinestérase et les antagonistes du récepteur N-méthyl-D-aspartate (NMDA). Sur la base de la littérature actuelle sur la recherche en pharmacologie de la maladie d’Alzheimer, l’analyse de gènes exprimés de manière différentielle dans des modèles médicament-médicament, maladie-maladie et médicament-maladie permet la découverte de nouveaux agents pharmaceutiques qui traitent potentiellement plus que les symptômes de la MA. Des outils analytiques tels que la carte de connectivité (cMap) ont été utilisés pour l’interaction médicament-maladie à partir de données de micropuces disponibles au public. Les signatures génétiques de l’interprétation basée sur cMap ont montré que les médicaments anti-AD courants (tacrine, donépézil, galantamine, mémantine et rivastigmine) ne figuraient pas dans la liste finale des médicaments. Au lieu de cela, d’autres composés qui inhibent les effecteurs en aval de la prolifération cellulaire, les voies Wnt et l’insuline, les modifications épigénétiques et la régulation du cycle cellulaire figurent parmi les premiers de la liste finale des médicaments anti-AD. Ces résultats ont également confirmé le fait que la MA est une maladie de dégénérescence et de dérégulation de la croissance. En fait, la liste finale des médicaments anti-AD, obtenue à partir de l’analyse de jeux de données de puces à ADN et du modèle médicament-maladie cMap, contenait l’effecteur commun de l’AD et du diabète – la glycogène synthase kinase 3 (GSK3), une enzyme qui s’est révélée être liée à l’hyperphosphorylation protéine tau) – a confirmé le lien entre les deux maladies. Une autre interprétation des gènes obtenus à partir de jeux de données de puces à AD utilisant KEGG, WikiPathways, Reactome, Biocarta et NetworkAnalyst a également montré que le facteur de croissance épidermique (EGF) et ses récepteurs étaient fortement associés à la pathogenèse de la MA. L’EGFR est une protéine transmembranaire et un membre de la famille des récepteurs HER / ErbB qui partagent une voie commune avec les récepteurs de l’insuline (Ras / Raf / Mak et PI3K / Akt). En outre, le précurseur de la protéine amyloïde (APP) était indirectement lié à l’analyse du réseau. Aβ (l’un des résultats diagnostiques de la MA) active l’EGFR et l’inhibition du récepteur améliore les troubles de la mémoire chez la drosophile Aβ surexprimée. Les médicaments qui bloquent GSK3 affectent la voie PI3K / Akt, démontrant que l’EGFR pourrait être une nouvelle cible pour l’agent pharmaceutique dans le traitement de la MA.